TransMedics’ OCS HEART delivers warm, oxygenated, nutrient-enriched blood to the donor heart and keeps it in a living state until the organ is ready to be transplanted. Image courtesy of TransMedics.


Latest News
May 2, 2016
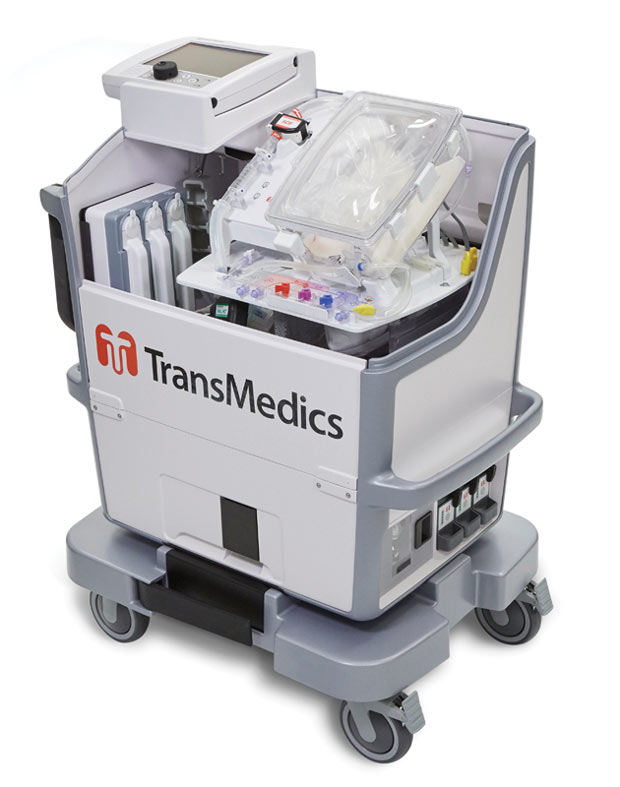 TransMedics’ OCS HEART delivers warm, oxygenated, nutrient-enriched blood to the donor heart and keeps it in a living state until the organ is ready to be transplanted. Image courtesy of TransMedics.
TransMedics’ OCS HEART delivers warm, oxygenated, nutrient-enriched blood to the donor heart and keeps it in a living state until the organ is ready to be transplanted. Image courtesy of TransMedics.When organ transplantation is your business, it’s critical to stay on the leading edge. TransMedics, known for its next-generation Organ Care System used to transport hearts, lungs and livers in a “living state” outside the body as opposed to the traditional biohazard ice cooler, struggled to strike a balance between innovating under time-to-market pressures and industry requirements around compliance.
After nearly a decade relying on mostly manual processes, TransMedics conducted an internal audit and found its compliance efforts in need of some serious resuscitation. The paper-based processes provided little visibility into design changes or approval workflows, and action items associated with specific projects would often get lost in a pile of documents or stuck in someone’s inbox. It was hardly a remedy for fast-tracking products through the FDA approval process and out to the open market.
“We decided that given the size of our company, the complexity of what we were developing, and our growth, it was time to move to an automated system,” says Progga Das, quality systems engineer at the medical device manufacturer. “You can achieve much higher visibility when you have online systems.”
TransMedics, like so many start-up medical device companies, found documenting repeatable processes to meet regulatory compliance to be more of a struggle than designing and engineering innovative products. Trying to meet requirements for regulations like the FDA 21 CFR Part 11, for electronic signatures, ISO standards for quality management and the CE Mark, to sell a product in Europe, is a complex drill that is often enforced and managed by personnel who aren’t facing the same time-to-market demands as engineers.
Meeting the compliance challenge involves an array of formal processes and procedures along with documentation and audit trails to prove everything has been kept in check. Manual processes, coupled with disparate design, quality assurance (QA) and homegrown systems, have been the norm. But with product complexity on the rise and a greater focus on quality, medical device makers are looking for a lifeline. They may have found it by automating compliance processes with document management systems and PLM (product lifecycle management).
“As an engineer, you always want to design the coolest products with the greatest feature set and you don’t want to waste time separating yourself from design to follow some due diligence process for compliance,” says Chuck Cimalore, CTO of Omnify Software, maker of Omnify Empower PLM. “The question is how to maximize time doing design work while retaining the confidence that you’re not stepping outside of the due diligence boundaries so you won’t run into regulatory issues downstream.”
Eliminating the Busy Work
Validation is the key to any compliance initiative. Document management systems provide numerous capabilities, including electronic audit trail, workflow and electronic signature functions, that ensure processes are repeatable and do so in a way that’s less time consuming and expensive than human oversight, says Todd Cummings, vice president of Research and Development at Synergis Software, the provider of the Adept engineering document management system. “We can take a lot of the details and busy work out of the process and automate it,” Cummings says. “That provides a massive leg up by allowing humans to focus on more creative thinking work.”
For example, version control, one of Adept’s features, keeps an audit trail of what documents or CAD models were checked in or out during the product lifecycle, ensuring everyone, not just engineers, are working off of the most current and accurate information, Cummings says. Electronic signatures, a critical piece of compliance directives, can also be streamlined in systems like Adept, through the use two-factor identification, which validates that a person signing off on something is who they say they are, he adds.
Quality, another big piece of compliance, can also be addressed more naturally within the context of a document management system, contends Greg Milliken, vice president of Marketing for M-Files, an enterprise document and content management system. As opposed to storing quality manuals on a shelf somewhere or in a separate siloed system where they are likely to be ignored, Milliken says moving that data and those processes under the domain of a document management system used every day is more likely to result in repeatable processes and as a result, compliance.
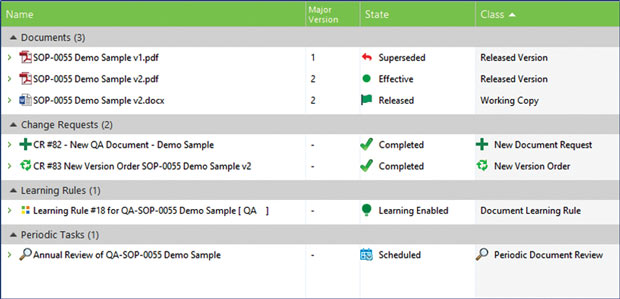 M-Files QMS includes built-in change request features, version history, audit trail and proofed evidence of document learning to ensure that employees adhere to policies related to compliance. Image courtesy of M-Files.
M-Files QMS includes built-in change request features, version history, audit trail and proofed evidence of document learning to ensure that employees adhere to policies related to compliance. Image courtesy of M-Files.“Our philosophy is to move away from a separate, siloed quality system to a system where it becomes day-to-day quality management,” he explains. “The first responsibility of maintaining quality is to document things that should be done to the best standards of the industry. Now imagine doing that within the same system that you’re doing everything else in.”
PLM and the Case for Quality
With the FDA shifting its compliance focus to quality, PLM vendors are making the case that their platforms are the best fit. Along with key industry partners, the FDA has been pushing the Case for Quality, an initiative launched in 2011 to identify and promote best practices for medical device quality after research revealed that the number of adverse event reports—particularly among cardiovascular and surgical devices—has increased significantly and is outpacing industry growth by a wide margin. The number of recalls on medical devices has also escalated, with a third due to design flaws and another quarter related to issues in manufacturing, the FDA found. Improving quality measures presents an enormous opportunity to the medical device industry—to the tune of about $4.75 billion to $6 billion, according to research from the McKinsey Center for Government.
Driven by Data
As opposed to a document-centric take on compliance, the new quality mandates require a data-driven approach, something only supported by PLM, notes Swapan Jha, PTC’s vice president of go-to-market for PLM. “When the design engineer or quality engineer is working on a 3D model of a medical device, a lot of data gets added to that model from a compliance and traceability perspective,” he explains. “Today, a lot of customers take that 3D model and create a PDF and all of that information that’s supposed to be tracked is lost and the dots are disconnected.”
With its model-based, product-centric view, Windchill PLM can provide access to the artifacts required for a quality initiative and for compliance throughout a product’s lifecycle, Jha says. With Windchill 11, PTC is making compliance easier by providing out-of-box configurations for certain regulations—CFR Part 820 for quality system regulation, for example, as well as several of the ISO standards.
 Aras PLM supports Failure Modes and Effects Analysis (FMEA) procedures to help identify potential failure modes within a system, process, design or item while helping to design those failures out with minimum time and resources. Image courtesy of Aras.
Aras PLM supports Failure Modes and Effects Analysis (FMEA) procedures to help identify potential failure modes within a system, process, design or item while helping to design those failures out with minimum time and resources. Image courtesy of Aras.For its part, Aras is touting its model-based environment as one of the primary reasons its PLM platform is gaining traction among medical device companies as a means to automate compliance, according to Doug Macdonald, the company’s product marketing director. The flexibility of Aras’ modeling approach for customizing business processes, coupled with PLM’s ability to serve as a digital thread, providing traceability from a product sitting in a warehouse all the way back through manufacturing and to design, makes it much more effective in streamlining compliance processes compared to any manual approach. “Without PLM, information could potentially be spread everywhere,” he explains.
A Layered Approach
Some Aras customers like Carestream Health Inc., are layering Aras on top of existing PLM systems to facilitate compliance and quality initiatives. Carestream, which manufactures medical imaging equipment among other health care-related offerings, uses Siemens PLM Software’s Teamcenter to manage engineering CAD and assembly files. However, the company has also brought in Aras PLM as the system of record for Device Master Records (DMR), which capture all the drawings, documents, work instructions and processing information related to a particular device, along with the Design History File (DHF), which captures the complete record of design decisions.
“Aras gives us more flexibility to work with from a configuration management perspective,” says David G. Sherburne, Carestream executive IT director. As for why PLM in general for compliance: “It helps us be more efficient while reducing compliance risk in a global environment—we want our cycles going into product innovation, not trying to figure out what version of the truth is out there,” he explains.
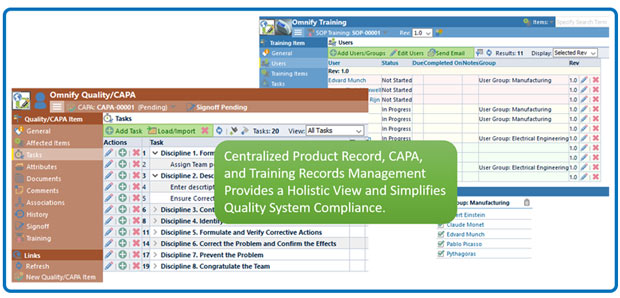 Omnify automates many quality and compliance processes, including training. Image courtesy of Omnify Software.
Omnify automates many quality and compliance processes, including training. Image courtesy of Omnify Software.For TransMedics, Omnify PLM was the prescription for balancing compliance requirements with the need to accelerate time to market. The platform automates many compliance processes, including QA training to ensure manufacturing operators stay up-to-date. “It’s critical that we make sure they have training before completing a specific process and we’re out of compliance if it’s not documented,” Das explains. “Training was one of the areas we thought we could improve by managing it in Omnify versus having someone remember what person was trained in what and losing all of that closure.”
Specialized Service Providers
While PLM and document management tools can automate and streamline compliance, they can’t do so without knowledge of the regulatory landscape, which continues to evolve. That’s where specialized service providers come into play. Sterling Medical Devices, for example, provides design and consulting services to medical device manufacturers, including in the area of compliance, which is only going to get more complex as medical device makers integrate state-of-the-art technologies like smartphones, the cloud and the Internet of Things (IoT) into their offerings.
The FDA is starting to adapt, but it’s a few years behind the technology, says Bruce Swope, vice president of Engineering at Sterling. “The FDA, ISO and CE bodies are constantly coming out with new versions of regulations,” Swope cautions. “You could be designing for old standards and if you’re not done in time when the new regulations are out, you won’t get approval if you don’t meet the new regulations.”
More Info
Subscribe to our FREE magazine, FREE email newsletters or both!
Latest News
About the Author
Beth Stackpole is a contributing editor to Digital Engineering. Send e-mail about this article to DE-Editors@digitaleng.news.
Follow DE